Publication: Genethon helps clarify a molecular mechanism of mitochondrial malfunction in Duchenne muscular dystrophy
Several laboratories, including Généthon, have begun gene therapy clinical trials for Duchenne muscular dystrophy. Research must be conducted, however, to improve the therapeutic benefit of future treatments for this rare genetic disease.
“In order to make further progress, we need to better understand the physiology of the disease,” explains David Israeli. “DMD is characterized by fragility of the muscle fibers, due to the lack or to the abnormal expression of the dystrophin protein, whose one of the primary functions is the mechanical stabilization of skeletal and cardiac muscles.” David explains that “dystrophin is a large and highly complex protein, with many molecular interactions and biological functions which go beyond its role as a mechanical stabilizer of myofibers.”
The aim of the research conducted by Ai Vu Hong, supervised by Isabelle Richard and David Israeli, is to clarify the mechanisms of mitochondrial malfunction in DMD.
For many years it was believed that disruption of calcium homeostasis was the principal cause of mitochondrial dysfunction in DMD.
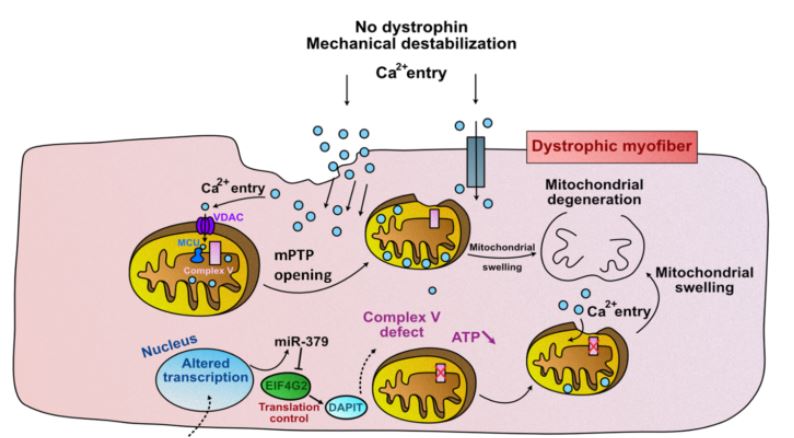
The results obtained in the study by Ai Vu Hong shed new light on this point. “We have recently found that in the DMD muscle, the upregulation of a microRNA (miR-379), represses the expression of mitochondrial proteins, and ATP production (Sanson et al., 2020). This reduction in energy production can accelerate the damage which is cause by the disruption of calcium homeostasis in the mitochondria of the dystrophic muscle”, David Israeli explains.
MiRNA-379, along with many other mi-RNAs, is part of the “Dlk1-Dio3 miRNAs cluster”, around twenty of which are deregulated in DMD (Jeanson-Leh et al.2014) Serum profiling identifies novel muscle miRNA and cardiomyopathy-related miRNA biomarkers in Golden Retriever muscular dystrophy dogs and Duchenne muscular dystrophy patients
Ai Vu Hong showed that, similarly to miRNA-379, other mi-RNAs from the same Dlk1-Dio3 cluster are also involved in the suppression of mitochondrial energy production.
He therefore proposes an updated model for mitochondrial malfunction in DMD (Figure 2).
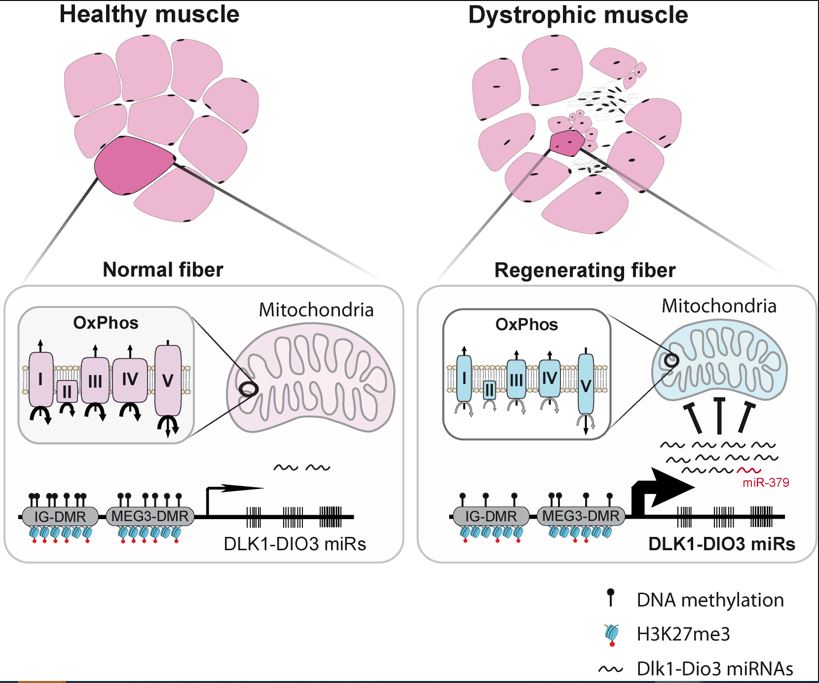
In summary, this publication contributes to the understanding of mitochondrial dysfunction in Duchenne muscular dystrophy.
Find out more
Vu Hong A, Bourg N, Sanatine P, Poupiot J, Charton K, Gicquel E, Massourides E, Spinazzi M, Richard I, Israeli D. October 21, 2022.